
Overview
|
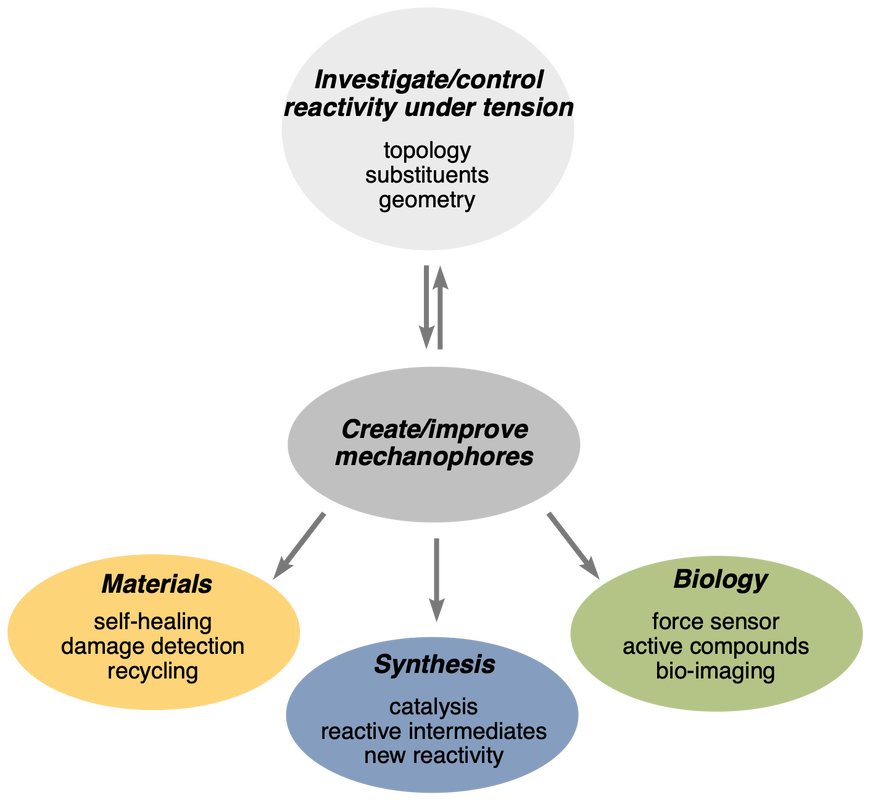
Mechanical force is a formidable, and relatively unexplored, source of energy that, with its ability to distort, bend and stretch chemical bonds, is unique in the way it activates chemical reactions. The precise control of this force could revolutionise how we build and rearrange molecular structures and change the way we think about chemical transformations. Although grinding is a powerful method to transmit mechanical energy and promote chemical reaction in the solid state, the energy is transmitted in non-specific and non-directional ways. More precise control can be achieved when the chemical entity that is the subject of the mechanical force (a “mechanophore”) is embedded within a polymeric backbone. Indeed, pulling both ends of a macromolecule apart creates highly directional strain with its highest intensity in the middle of the chain. Our research aims at controlling mechanical force at the molecular level for application in synthetic chemistry, materials and biology. This approach relies on a varied set of skills: synthetic organic and polymer chemistry, supramolecular chemistry and molecular modelling.
Learn more about our methodology. |
|
Recent projects
|
For an overview of our research, watch Guillaume's webinar for the CMCC YouTube channel.
|
|
Force-controlled release
|
Force-controlled release of small molecules offers great promise for the delivery of drugs and the release of healing or reporting agents in a medical or materials context. In polymer mechanochemistry, polymers are used as actuators to stretch mechanosensitive molecules. This technique has enabled the release of molecular cargo but the systems described so far have been limited in the diversity and/or quantity of the molecules released per stretching event. This is due to the difficulty in iteratively activating scissile mechanophores, as the actuating polymers will dissociate after the first activation. We have shown that a rotaxane acts as an efficient actuator to trigger the release of cargo molecules appended to its axle, including representative functional molecules (a drug, a fluorescent tag and an organocatalyst). We anticipate that a large variety of cargo molecules could be released with this device. Read the paper.
|
|
Controlling reactivity under tension
The activity of a mechanophore can be controlled by playing with electronics, gating, and stereo-, regio-, or even topochemical effects.
|
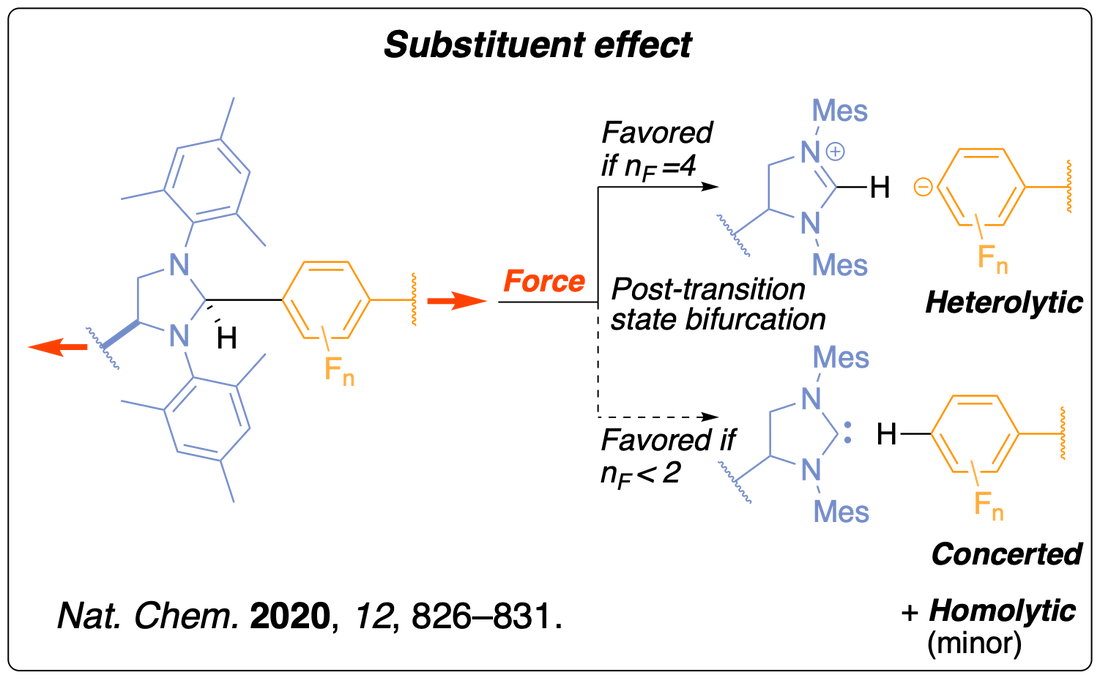
We have uncovered the existence of three concomitant dissociation pathways during the mechanical activation of a N-heterocyclic carbene (NHC) precursor and found that the products distribution could be controlled with the polarisation of the scissile bond. We determined that the selectivity is the result of the post-transition-state bifurcation on the force-modified potential energy surface. NHCs are versatile catalysts and our mechanophores could be used for the development of mechanocatalysts to trigger self-healing or facilitate recycling in/of materials. Read the paper.
|
|
|
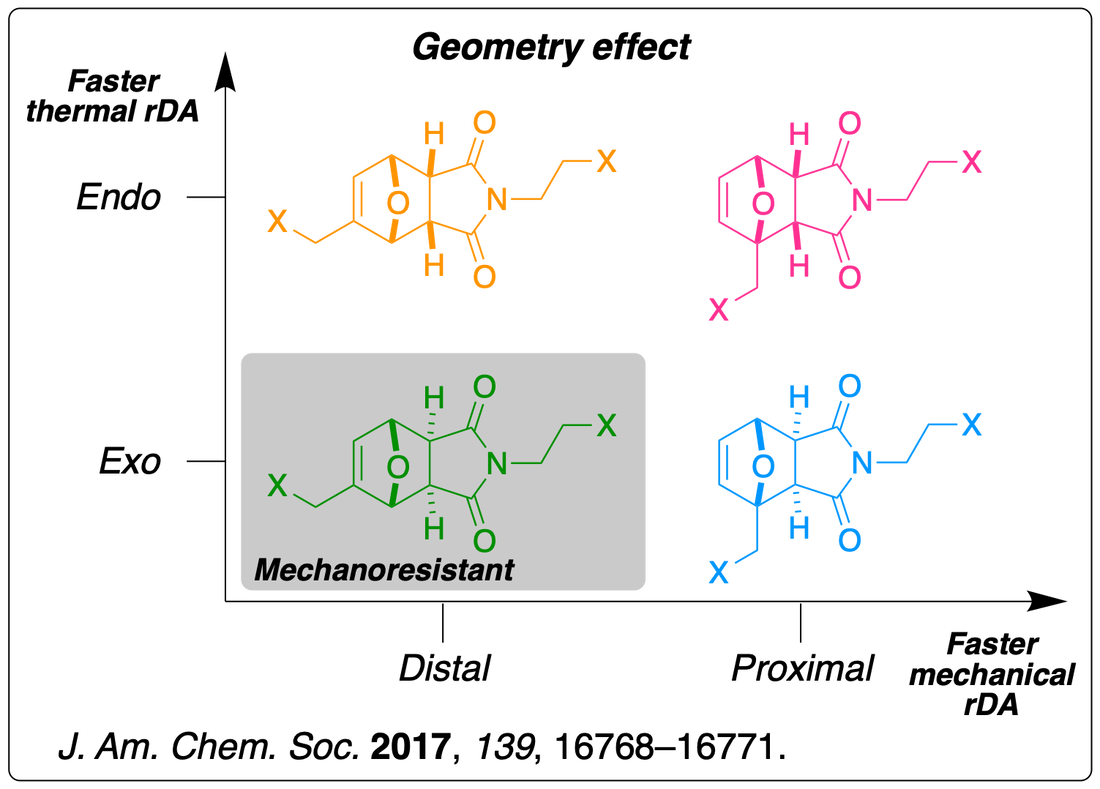
We have investigated the force-accelerated retro-Diels-Alder reactions of furan/maleimide adducts, and found that the mechanochemical reactivity can be enhanced or completely suppressed depending on the regio- and stereochemistry of the mechanophores. These units are popular cross-linkers in thermally-mendable materials and our finding could be used to finely tune the thermo-mechanical behaviour of such materials. Read the paper.
|
|
Mechanochemistry of the mechanical bond
Mechanical bonds, with their capacity for large amplitude movements, are ideally suited to interface mechanical force with mechanophores, but these interlocked structures are themselves mechanically active and susceptible to degradation. Read our review.
|
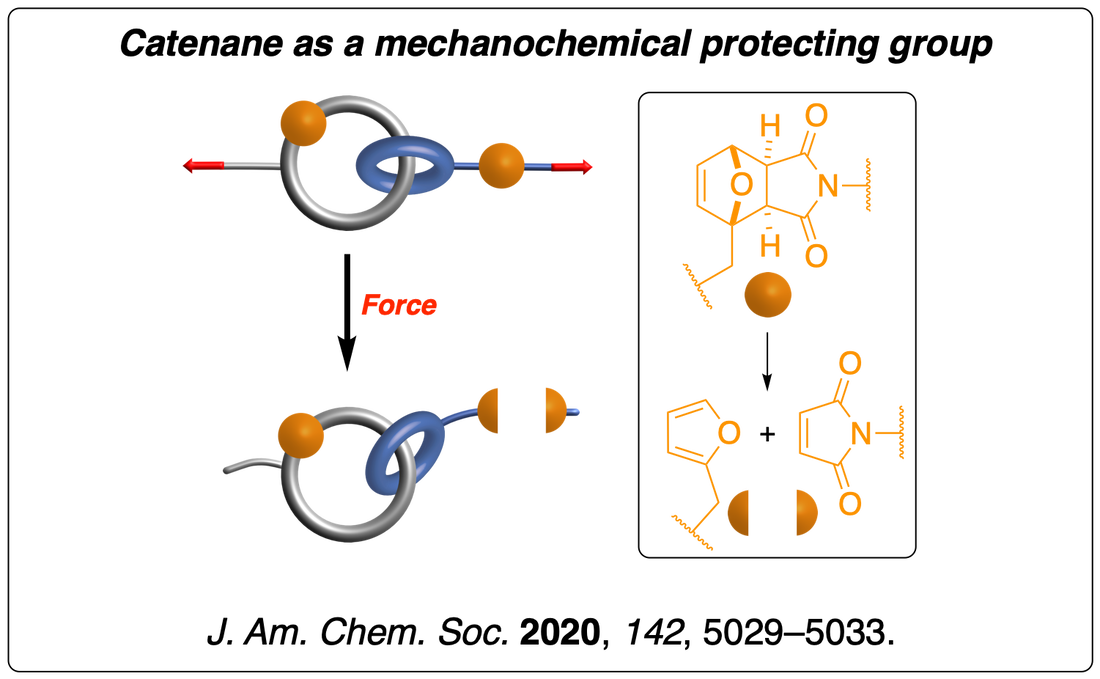
We have shown that a catenane acts as a mechanical protecting group by spreading the tensile deformation across the entire framework of the catenane, thereby protecting a mechanophore incorporated in one of its rings. This approach provides a new way to control the mechanical activity of a mechanophore. Read the paper.
|
|
|
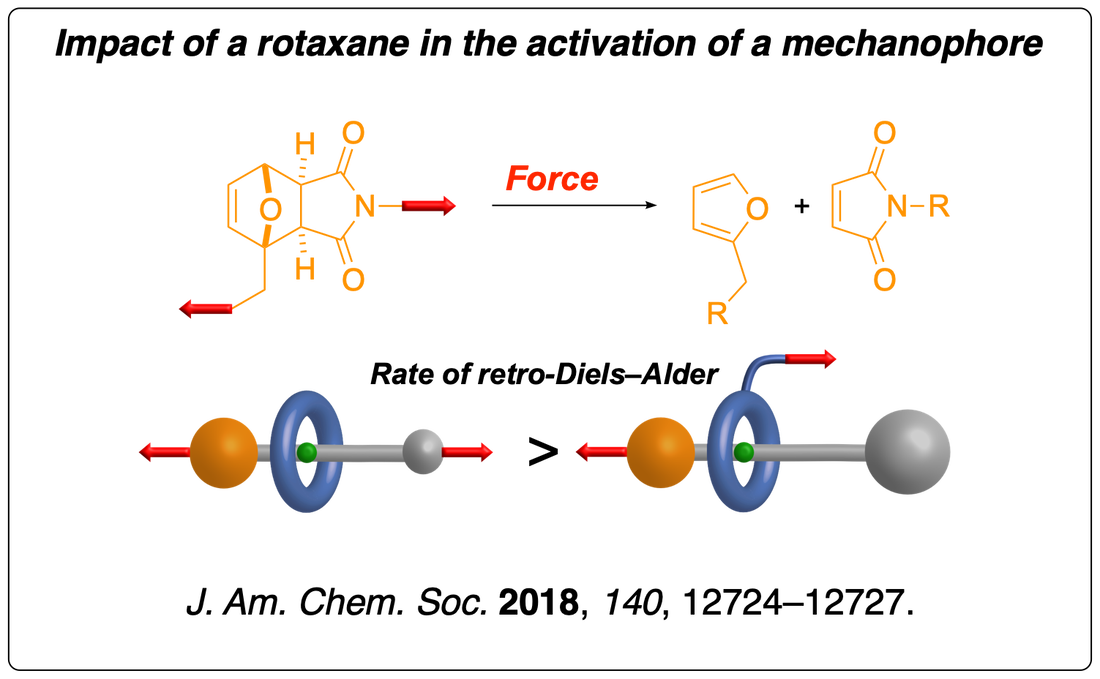
We have shown that a rotaxane architecture could delay the activation of a mechanophore embedded in its axle due to the emergence of a competing high-stress region where the axle is constricted by the macrocycle. Read the paper.
|
|
|
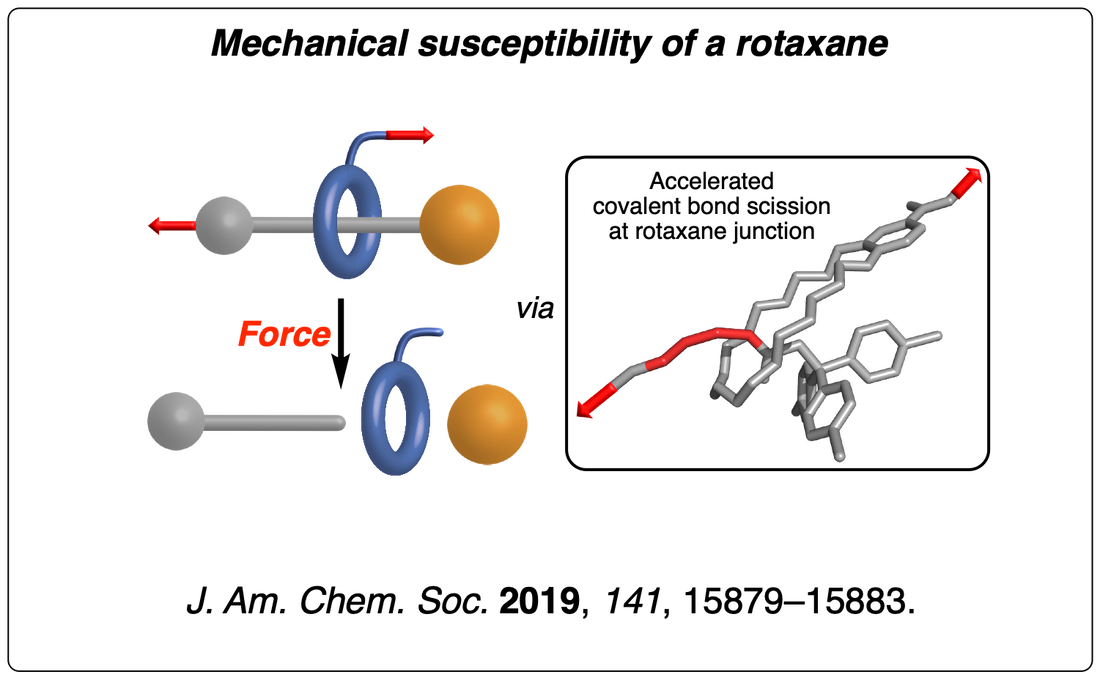
We have then found that the same high-stress region can lead to the selective cleavage of a covalent bond in the axle in the absence of a competing mechanophore. These results suggest that the rotaxane architecture acts as a lever that accelerates the dissociation of interlocked covalent bonds by lowering the total loading force required to provoke their rupture. This finding has significant implications for the future development of slide-rings materials (a network of polyrotaxanes connected by their macrocycles). They display a remarkable ability to absorb mechanical energy at low extension due to “pulley effect” of their mobile cross-links. However, our results suggest that their rotaxane architecture could accelerate the failure of this material at maximal elongation (when the macrocyclic cross-links are forced against the terminal stoppers of the polymer axles). Read the paper.
|
|